Мукополисахаридоз I типа

Медицинский эксперт по заболеванию
Проф. Полина СтепенскиРуководитель отделения ТКМ и иммунотерапии у детей и взрослых клиники «Хадасса»
Стаж: более 24 лет
Запись на прием
В Израиле прекрасных результатов при лечении мукополисахаридоза 1 типа добивается всемирно известный специалист по трансплантации костного мозга профессор Полина Степенски, заведующая отделением ТКМ и иммунотерапии клиники «Хадасса», член Европейского комитета по ТКМ при не злокачественных заболеваниях.
В клинике также очень эффективно применяют фермент-заместительную терапию. Ведущие эксперты в области детской кардиологии, ортопедии, неврологии, пульмонологии и многих других областях медицины разрабатывают эффективные индивидуальные программы паллиативной терапии.
Что такое мукополисахаридоз 1 типа
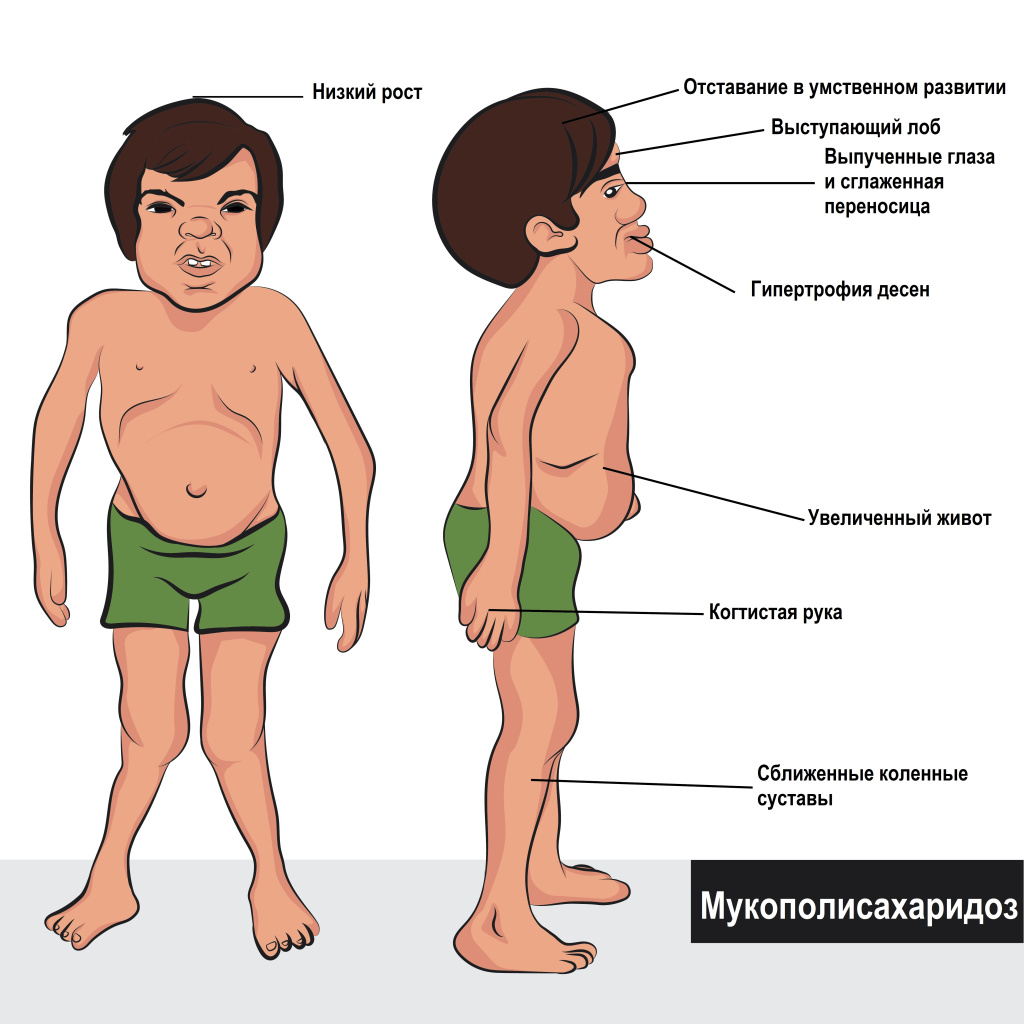
Мукополисахаридоз 1 является генетическим орфанным заболеванием с аутосомно-рецессивным типом наследования, при котором нарушен обмен веществ. Мукополисахаридоз 1 (МПС-1) — лизосомная болезнь накопления кислых мукополисахаридов в тканях различных органов. Распространенность болезни в Европе составляет 1:200, в России 1 случай на 400 тыс. новорожденных. При мукополисахаридозе 1 типа поражается ЦНС, костно-суставная, дыхательная и сердечнососудистая система, а также печень и селезенка, зрение и слух. Синдром Гурлер-Шейе — наиболее тяжелая форма среди 11 типов мукополисахаридозов, отличается быстрым прогрессированием и низкой продолжительностью жизни.
Существует 3 клинических варианта мукополисахаридоза 1 типа: синдром Гурлер (MPS-I H), синдром Шейе (MPS-I S)и синдром Гурлер-Шейе (MPS-I H/S). Все три вида заболевания отличаются характером течения и симптоматической картиной. Наиболее тяжелая и часто встречающаяся форма МПС — синдром Гурлер, впервые описанного австрийским педиатром Г. Гурлер. Синдром Шейе — мягкая форма мукополисахаридоза 1 типа, а синдром Гурлер-Шейе характеризуется промежуточной степенью тяжести. В популяции независимо от географического фактора мукополисахаридоз 1 типа встречается приблизительно в 1 случае на 100 тыс. живорожденных младенцев. Заболевание обусловлено мутациями в гене лизосомного фермента альфа-L-идуронидазы, недостаточность которого приводит к накоплению мукополисахаридов в соединительной ткани и существенному повреждению различных органов.
Код по МКБ-10.
По международной классификации болезней десятого пересмотра мукополисахаридоз 1 типа имеет код Е76.0.
Чтобы получить более подробную информацию или консультацию специалиста по орфанным заболеваниям, заполните все поля формы. Наши консультанты будут рады Вам помочь.
Патогенез
Мукополисахаридоз 1 имеет аутосомно-рецессивный тип наследования, поэтому с одинаковой частотой встречается как у мальчиков, так и у девочек. Если от матери и отца ребенок получает обе дефектные аутосомы по данному гену, мукополисахаридоз 1 типа клинически манифестирует. Вероятность наследования болезни составляет 25 %, если оба родителя несут мутантный ген. Мукополисахаридоз 1 типа может дебютировать в младенчестве или подростковом возрасте. При заболевании нарушается функция лизосомных ферментов, в результате чего накапливаются гликозаминогликаны. В результате, патологический процесс приводит к гибели клеток и тканей различных органов и систем.
Симптомы и проявления
Мукополисахаридоз 1 типа — мультисимптомное заболевание. Начало манифестации, характер и выраженность проявлений зависят от клинической формы болезни. Врачи первичного звена во время осмотра ребенка могут заподозрить мукополисахаридоз 1 типа по основным признакам, таким как: костные деформации без воспалительных проявлений (контрактура, грудопоясничный кифоз), повторные возникновения грыжи, рецидивные отиты и респираторные заболевания, недостаточность сердечных клапанов, увеличение печени и селезенки. Если у ребенка были выявлены 3-4 подобных симптома, необходимо обратиться к генетику.
Клиническая картина
Три варианта мукополисахаридоза 1 типа отличаются следующими особенностями и проявлениями:
Синдром Гурлер (MPS-I H)
Синдром Гурлер (MPS-I H) — характеризуется быстрым, прогрессирующим течением и тяжелой симптоматикой, встречается в 1 случае на 25 тыс. новорожденных. В первые месяцы жизни младенцы с синдромом Гурлер развиваются нормально. Специфические признаки болезни возникают ближе к годичному возрасту. В большинстве случаев к 1-2 годам формируется полная клиническая картина. Синдром Гурлер проявляется отставанием в росте, прогрессирующими деформациями скелета, в том числе искривлением позвоночника, недоразвитием костей лицевого черепа, дисплазией тазобедренных суставов, нижнепоясничным лордозом.
Мукополисахаридоз 1Н типа (синдром Гурлер) сопровождается гепатомегалией, спленомегалией, макроцефалией, краниосиностозом, пахово-мошоночными и пупочными грыжами, нарушением координации движений, параличом, парезами, диффузным поражением миокарда, заболеваниями слухового и зрительного аппарата. Также у детей с синдромом Гурлер отмечаются: избыточный рост пушковых волос, своеобразная походка (на полусогнутых ногах), изменения сухожильных рефлексов и мышечного тонуса, умственная отсталость, задержка речевого развития, внешние пороки развития области носа и лица.
Синдром Шейе (MPS-I S)
Синдром Шейе (MPS-I S) — отличается относительно благоприятным течением, до 3-6 лет обычно никак не проявляется. Полная клиническая картина синдрома Шейе формируется до подросткового возраста. Первые симптомы при мукополисахаридозе 1S типа: контрактуры суставов верхних конечностей. У детей с синдромом Шейе наблюдается гипертрихоз, частое возникновение грыж, незначительное увеличение селезенки и печени. По мере прогрессирования мукополисахаридоза 1S типа отмечается нистагм, развивается глаукома, дистрофия сетчатки, помутнение роговицы, что нередко приводит к потере зрения. При синдроме Шейе интеллектуальные способности в норме. Не исключены аортальные пороки сердца. Присутствуют признаки гаргоилизма: грубые черты лица, большой рот и язык, широкие скулы, толстые губы, крупный череп, запавшая переносица.
Чтобы получить более подробную информацию или консультацию специалиста по орфанным заболеваниям, заполните все поля формы. Наши консультанты будут рады Вам помочь.
Синдром Гурлер — Шейе (MPS-I H/S)
Синдром Гурлер — Шейе (MPS-I H/S) — промежуточная форма между MPS-I H и MPS-I S, характеризуется умеренной, более мягкой клинической картиной, высокой продолжительностью жизни. Частота встречаемости 1 случай на 80-100 тыс. новорожденных. Манифестация синдрома Гурлер-Шейе начинается у детей в возрасте 1-2 лет. Мукополисахаридоз 1 H/S типа сопровождается обструкцией верхних дыхательных путей, поражениями сердца, а также тотальным спондилолистезом (смещением нижележащего позвонка), что нередко приводит к компрессии спинного мозга. У пациентов с синдромом Гурлер-Шейе незначительно сниженный интеллект, поэтому они нормально учатся и получают образование в любом учебном учреждении.
Диагностика
По совокупности симптомов и внешних признаков врачи могут заподозрить у ребенка один из фенотипов мукополисахаридоза 1 типа еще в раннем возрасте. Для подтверждения диагноза назначают комплексное обследование, включающее такие процедуры:
- биохимический анализ мочи на количество мукополисахаридов;
- исследование крови для определения уровня альфа-L-идуронидазы в клетках крови;
- ДНК-анализ, подтверждающий генетические дефекты;
- рентгенография скелета;
- МРТ, УЗИ различных органов и систем.
Также пациентам требуется эхокардиография и осмотр специалистов различных направлений: психиатра, отоларинголога, офтальмолога, невролога, кардиолога и др.
Лечение
Выбор терапевтических методов при мукополисахаридозе I типа зависит от формы заболевания, степени тяжести клинической картины и возраста пациента. Программа лечения направлена на улучшение качества и увеличение продолжительности жизни.
Консервативное лечение — заключается в использовании противовоспалительных и сердечно-сосудистых препаратов, нейрометаболических стимуляторов, витаминов А, В, Е, С и фолиевой кислоты, гепатопротекторов. Также в курс консервативного лечения синдрома Гурлера-Шейе и других форм болезни входит ферментозаместительная терапия — внутривенное введение недостающего фермента альфа-L-идуронидазы. Кроме того, при мукополисахаридозе I типа показаны физиотерапевтические процедуры: магнитотерапия, лазерная акупунктура, ЛФК, электрофорез, массаж и т.д.
Хирургическое лечение — включает в себя иссечение грыж, протезирование тазобедренного сустава, операционные вмешательства на сердце (замена клапанов). В некоторых случаях требуется декомпрессия спинального канала, шунтирование гидроцефалии, трахеостомия и аденотонзиллэктомия (удаление миндалин и аденоидных тканей). Также проводятся антиглаукоматозные операции.
Трансплантация костного мозга — введение здоровых донорских клеток, способных синтезировать необходимый фермент. Процедура позволяет остановить прогрессирование мукополисахаридоза I и тем самым улучшить качество жизни пациентов.
Возможные осложнения и прогноз
Мукополисахаридоз I типа осложняется респираторными инфекциями, обструкциями дыхательных путей, сердечной недостаточностью. Синдром Гурлер имеет неблагоприятный прогноз. Дети с этой формой заболевания на фоне тяжелых патологических изменений многих органов и систем не доживают до 10 лет. При мукополисахаридозе I S и H/S в большинстве случаев при условии качественного симптоматического лечения складывается благоприятная прогностическая картина. Пациенты с такими клиническими формами мукополисахаридоза I получают образование, социализируются, создают семьи и живут полноценной жизнью.
Если вам необходима консультация специалиста по орфанным заболеваниям, заполнив все поля формы, и наши консультанты будут рады Вам помочь.
https://rare-disease.ru/diseases/mukopolisaharidoz-i-tipa/